

使用 SP3 純化基于凝膠的樣品分級用于自上而下的蛋白質組學
Gel-Based Sample Fractionation with SP3-Purification for Top-Down Proteomics
Ayako Takemori, Naoyuki Sugiyama, Jake T. Kline, Luca Fornelli, and Nobuaki Takemori
Journal of Proteome Research 2025 24 (2), 850-860
DOI: 10.1021/acs.jproteome.4c00941
摘要:蛋白質組樣品的精確預分餾是在自上而下的蛋白質組學中實現深入分析的有效方法。PEPPI-MS(被動洗脫聚丙烯酰胺凝膠中蛋白質作為 MS 的完整物質)是一種基于凝膠的樣品分級分離方法,通過在 SDS-PAGE 分離后從聚丙烯酰胺凝膠中高效提取蛋白質,實現基于分子量的高分辨率蛋白質組分離。此后,在質譜分析之前,必須有效地去除 PEPPI 餾分中的 CBB 和 SDS 等污染物。在這項研究中,通過將 PEPPI-MS 與 SP3(單罐固相增強樣品制備)中使用的基于磁珠的蛋白質純化方法相結合,我們開發了一種完整、穩健且簡單的樣品制備工作流程,用于自上而下的蛋白質組學在 PEPPI-SP3 中,從凝膠中提取的蛋白質收集在 SP3 珠子的表面,用有機溶劑洗滌,并用含有 0.05% (w/v) SDS 的 100 mM 碳酸氫銨完整回收。使用陰離子交換 StageTip 進行額外純化后,對回收的蛋白質進行質譜分析。使用人細胞裂解物進行的性能驗證顯示,與使用有機溶劑沉淀或超濾的常規 PEPPI 工作流程相比,低分子量蛋白質回收率顯著提高,變異系數更低。
SP3磁珠請參考 http://www.posuichina.com/Product/1736805216.html
介紹
單個基因在體內產生具有不同化學結構的各種翻譯產物,稱為蛋白質形式。蛋白形式有助于蛋白質相互作用和亞細胞定位的多樣性,從而擴展蛋白質的生理功能。雖然蛋白質形式的總數仍然未知,但據估計,該數量比人類基因的數量22,800 個高出 100 多萬個 。了解龐大的人類蛋白質組學是當今蛋白質組學研究的目標之一,為實現這一目標,迫切需要開發全面的蛋白質組分析技術。
自下而上的蛋白質組學 (Bottom-up proteomics, BUP) 是人類蛋白質組大規模分析的主要分析方法,用于分析通過酶消化獲得的肽片段,原則上通常難以應用于蛋白質形式的高度準確鑒定。因此,允許直接分析完整蛋白質形式的自上而下的蛋白質組學 (Top-down proteomics, TDP) 被用作用于此目的的主要分析方法。在 TDP 分析中,樣品制備的影響不容小覷,正如 Kaulich 等人最近的一份報告中所詳細描述的那樣。蛋白質形態鑒定通常是通過反相液相色譜 (LC) 或毛細管電泳 (CE) 分離完整的蛋白質形態,然后在線連接質譜儀中碎裂來實現的。然而,為了從生物樣品中檢測出更多的蛋白質組分,在 LC/CE 分離之前進行樣品預分餾是必不可少的。
SDS-聚丙烯酰胺凝膠電泳 (PAGE) 是生化實驗中的一種核心蛋白質分離方法,通過在聚丙烯酰胺凝膠中添加陰離子表面活性劑 SDS,根據分子量 (MW) 對線性化蛋白質進行電泳分離。SDS-PAGE 的特性能夠對細胞裂解物中的復雜蛋白質組進行高分辨率分離,并被廣泛用作 BUP 中一種強大的樣品預分餾方法。2020 年,我們開發了 PEPPI-MS,這是一種用于被動提取凝膠中蛋白質的高效方法,并成功地顯著提高了凝膠中蛋白質的回收率。盡管被動提取通常簡單且具有成本效益,但它也與低回收率、長提取時間和難以應用于高 MW 蛋白質有關。PEPPI-MS 通過使用考馬斯亮藍 (CBB) 和 SDS 作為提取增強劑解決了這些缺點,即使對于高 MW 蛋白,也能在 10 分鐘內實現高回收率。
在 PEPPI-MS 中,蛋白質組按如下方式分級分離:(1) 通過 SDS-PAGE 分離后在凝膠中添加 CBB,(2) 切除感興趣 MW 區域的樣品泳道,(3) 將凝膠塊搗碎并在含有 0.05–0.1% (w/v) SDS 的 100 mM 碳酸氫銨溶液 (pH 8) 中被動萃取 10 分鐘。由于獲得的餾分含有 CBB 和 SDS, 這會干擾 MS 分析,因此蛋白質純化是必不可少的,目前使用甲醇-氯仿-水沉淀 (MCW) 或陰離子交換轉盤輔助順序樣品制備 (AnExSP)。MCW 適用于約 300 μL 的小體積溶液,例如 PEPPI 餾分,是一種廉價且簡單的蛋白質純化方法。AnExSP 是一種使用陰離子交換固相萃取 (SPE) 微量離心柱(稱為 AX-StageTip)從樣品中去除 SDS 和 CBB 的方法。AnExSP 允許純化 PEPPI 組分,而不會損失低 MW 蛋白,這通常是 MCW 的一個問題。在 AnExSP 中,CBB、SDS 和 PEPPI 餾分中的蛋白質最初被捕獲在 StageTip 的陰離子交換 SPE 盤上,在隨后的洗脫過程中,使用乙醇和/或含有甲酸 (FA) 的乙腈溶液回收蛋白質。然而,標準 300 μL PEPPI 餾分中的 SDS 量超過了 AX-StageTip 可能去除的限度,需要事先進行 FASP 處理,這是一種使用離心超濾裝置進行繁瑣的尿素清洗。
在本研究中,我們嘗試開發一種使用 SP3 進行 PEPPI 分級分離的新純化方法,SP3 是目前 BUP 的主流樣品預處理方法。 在 SP3 中,通過添加有機溶劑(如乙醇或乙腈)沉淀的蛋白質被吸附到羧酸鹽修飾的磁珠表面,回收的蛋白質直接在磁珠上進行酶消化。通過可靠、簡單的作選擇性回收蛋白質,可有效去除 SDS 和其他污染物,其特點是在處理痕量蛋白質樣品時具有很高的重現性。結合最近開發的用于多樣品高通量處理的商業自動化系統,該方法可應用于多樣品處理。SP3磁珠請參考 http://www.posuichina.com/Product/1736805216.html
盡管 SP3 具有出色的樣品純化特性,但目前其在 TDP 中的應用受到限制,部分原因是難以回收吸附在磁珠上的完整蛋白質。Webb 小組的一項開創性研究報告稱,在 ?80 °C 下用 80% (v/v) FA 孵育可有效從磁珠中回收完整蛋白質,但條件對于高通量應用來說太苛刻了。 為了使 SP3 適應 TDP,我們開發了新的實驗條件,允許在室溫下從磁珠中快速且可重現地回收蛋白質,從而為TDP 提供了一種簡化而高效的樣品分離工作流程,該工作流程結合了基于 PEPPI 的蛋白質組分分離和基于磁珠的餾分純化,稱為 PEPPI-SP3。PEPPI-SP3 實現的基于 MW 的高分辨率蛋白質組分離可最大限度地減少低 MW 蛋白的損失,從而實現高通量樣品制備、全面分析,并最終實現多樣品處理。
實驗部分
補充方案中給出了先前發布的程序(如樣品制備、SDS-PAGE、PEPPI 分級分離和蛋白質純化)的詳細方案。
材料
除非另有說明,否則用于 PEPPI 分級分離和蛋白質純化的試劑購自 Wako(日本大阪),用于 TDP 分析的試劑購自 Fisher Scientific(美國伊利諾伊州羅克福德)。
細胞樣品
本研究使用了 MS 相容的人蛋白提取物 (Promega, Madison, WI, USA),一種市售的人類細胞蛋白提取物 (HCPE)。在 SDS-PAGE 之前,通過將 100 μL HCPE(1 mg 蛋白質)與 1 μL 500 mM 二硫蘇糖醇 (DTT) 在 37 °C 下孵育 90 分鐘進行還原處理,然后通過在 23 °C 下與 1 μL 1 M 碘乙酰胺在黑暗中孵育 30 分鐘進行烷基化處理。在 Amicon 離心式 3 kDa 超濾裝置(Merck Millipore,達姆施塔特,德國)中用 0.05% (w/v) SDS/100 mM 碳酸氫銨 (ABC) 替換溶劑,并調節至 2 μg/μL 的蛋白質濃度。
SDS-PAGE
使用 4–12% 預制凝膠 NuPAGE bis-tris(1 mm 厚,10 孔)和 NuPAGE MES 電泳緩沖液(Thermo Fisher Scientific,Waltham,MA,USA)進行 SDS-PAGE。將樣品與 NuPAGE LDS 上樣緩沖液 (Thermo) 混合,然后上樣到凝膠孔中。在 180 V 的恒定電壓下進行電泳后,從凝膠盒中取出凝膠,用 EzStain AQua(ATTO,Tokyo,Japan)染色 8 分鐘,然后用去離子水洗滌 30 分鐘至 1 小時。對于 TDP,進行了以下調整:使用自制的 10% 凝膠(1 mm 厚,10 孔)代替 NuPAGE 4–12% 凝膠(1 mm 厚,10 孔),并使用 Bio-Safe 考馬斯染色劑(Bio-Rad,Hercules,CA)染色 60 分鐘,而不是 EzStain AQua 染色 8 分鐘。
PEPPI 分級分離
使用 MW 標記作為指示劑,用工藝刀在感興趣的 MW 區域對染色凝膠的樣品泳道進行切片。將切除的凝膠塊收集在 BioMasher II 管(Nippi,Tokyo,Japan)中,并用塑料杵精細研磨。將凝膠進一步與 250 μL 0.05% (w/v) SDS/100 mM ABC 混合,并在管式混合器中以 23 °C、1500 rpm 搖動 10 分鐘。使用離心過濾器去除凝膠,所得溶液(約 250 μL),即 PEPPI 組分,用于后續的蛋白質純化:MCW、FASP 或 SP3。
MCW
將 PEPPI 餾分轉移至 1.5 mL 微管中,加入 600 μL 甲醇、150 μL 氯仿和 400 μL 超純水,混合,并在 23 °C 下以 13,500 rpm 離心 3 min。 離心形成雙層溶液,其中去除上層,向剩余的底層加入 400 μL 甲醇,并在 23 °C 下以 13,500 rpm 離心 3 分鐘。 用 400 μL 甲醇洗滌所得蛋白質沉淀物,并風干 30 分鐘。
FASP
將 PEPPI 餾分轉移至 Amicon 離心式 3 kDa 超濾裝置中,然后用 8 M 尿素置換溶劑,再用 100 mM ABC 置換溶劑。如前所述,通過 AnExSP(自制 AX-StageTip)純化裝置中的溶液:(11) 使用前用甲醇洗滌 AX-StageTip 并用 100 mM ABC 平衡;將樣品上樣至 AX-StageTip 上,用 40 μL 100 mM ABC 洗滌,用 40 μL 0.5% (v/v) FA/50% (v/v) 乙醇洗脫,再用 40 μL 0.5% (v/v) FA/50% (v/v) 乙腈洗脫,最后三個步驟分別以 7000 rcf 離心 3 分鐘。在 LC-MS 分析之前,將所得洗脫液在離心蒸發器中干燥。
SP3
SP3磁珠請參考 http://www.posuichina.com/Product/1736805216.html。將購自 Cytiva(美國馬薩諸塞州馬爾堡)的 Sera-Mag SpeedBead 羧酸鹽修飾的 E7 磁珠(50 mg/mL,貨號 45152105050250)和 E3 磁珠(50 mg/mL,貨號 65152105050250)各 50 μL 添加到 1.5 mL 微管中。用 800 μL 超純水洗滌 3 次后,將珠子懸浮在 250 μL 超純水中并儲存在 4 °C 直至使用。將先前制備的 PEPPI 組分轉移至 1.5 mL 微管中,并與 25 μL SP3 珠懸浮液 (20 μg/μL) 和 1.2 mL 乙醇混合;在 23 °C 下以 1200 rpm 振蕩 10 分鐘后,將微管置于磁力架上 3 分鐘,并通過抽吸除去液體。用 800 μL 80% (v/v) 乙醇洗滌磁珠兩次,用 800 μL 乙腈洗滌一次。為了回收吸附在磁珠上的完整蛋白質,在 23 °C 下用 20 μL 0.05% (w/v) SDS/100 mM ABC 在微管中以 2000 rpm 振蕩磁珠 5 分鐘。 將微管置于磁力架上 3 分鐘,并如上所述在 fasp 后半部分對含有回收蛋白質的溶液進行 AnExSP 純化。
ContamSpot 檢測
通過 ContamSpot 測定法測定純化后 PEPPI 組分的殘留 SDS 濃度。(19) 將純化的 PEPPI 餾分溶于 10 μL 0.1% (v/v) FA/2% (v/v) 乙腈中,用于測定。將 2 μL 樣品、2 μL 0.1% (w/v) 鄰甲苯胺藍和 5 μL 乙酸乙酯混合在 0.2 mL PCR 管中,并以 2000 rpm 離心 15 秒。從通過離心分離成兩層的溶液中,將 1.5 μL 乙酸乙酯層點樣到薄層色譜 (TLC) 板上。
胰蛋白酶/Lys-C 消化
在 LC-MS 分析之前,使用 RapiGest(沃特世,美國馬薩諸塞州米爾福德)溶解 PEPPI 餾分,并使用 MS 級胰蛋白酶/賴氨酸-C 混合物 (Promega) 消解。將來自不同 4 個 MW 區域的純化 PEPPI 組分溶于 10 μL 0.1% (w/v) RapiGest/100 mM ABC 中,并混合在 1.5 mL 微管中。將混合級分(總共 40 μL)與 0.2 μg 胰蛋白酶/賴s-C 混合物混合,并在 37 °C 下消化 16 小時。 為了降解 RapiGest,向消解的樣品中加入 10 μL 2.5% (v/v) 三氟乙酸 (TFA),并在 37 °C 下孵育 30 分鐘。 在 23 °C 下以 13,500 rcf 離心 10 分鐘后,通過自制的 SDB-StageTip 純化上清液并進行 LC-MS 分析。
消化肽的 NanoLC/MS/MS
在 timsTOF Pro2 質譜儀(Bruker Daltonics,Bremen,Germany)上使用 CaptiveSpray 納離子噴霧源 (Bruker Daltonics) 結合 nanoElute 2 納流 UHPLC 系統 (Bruker Daltonics) 獲取 MS 和 MS/MS 數據。在 50 °C 下,使用 PepSep ULTRA C18(250 mm × 75 μm、1.5 μm,Bruker Daltonics)作為分析柱。 將干燥的肽混合物溶于 30 μL 0.1% (v/v) TFA 和 5% (v/v) 乙腈中,每次進樣使用 2 μL 溶液進行 LC/MS/MS。流速為 400 nL/min,流動相由 (A) 0.1% (v/v) FA 的水溶液和 (B) 0.1% (v/v) FA 的乙腈溶液組成。采用多級線性梯度:流動相B在60 min內從4%增加至20%,在30 min內從20%增加至28%,在15 min內從28%增加至40%,在5 min內從40%增加至100%,并在100% B下保持10 min。
timsTOF 在 PASEF (parallel accumulation and serial fragmentation) 模式下運行。施加 1500 V 的毛細管電壓、3.0 L/min 的干燥氣體和 180 °C 的干燥溫度。MS 和 MS/MS 掃描范圍為 m/z 100–1700,1/K0 范圍為 0.6 至 1.6 Vs/cm2,斜坡時間為 100 ms,累積時間為 100 ms。碰撞能量根據離子淌度線性遞增,從 1/K0 = 1.60 Vs/cm2 的 59 eV 到 1/K0 = 0.60 Vs/cm2 的 20 eV。對于數據依賴性采集 (DDA) 分析,每個周期進行 1 次 MS 掃描和 10 次 PASEF MS/MS 掃描,預定目標強度為 20,000。使用多邊形過濾器,根據 m/z 和離子淌度選擇單電荷和低 m/z 離子,從源離子母離子選擇中排除。主動排除時間設置為 0.4 min。對于數據非依賴型采集 (DIA) 分析,DIA 窗口設置如表 S1 所示。MS 原始數據和分析文件已通過 jPOST 合作伙伴存儲庫 (https://jpostdb.org) 存放到 ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org),數據集標識符為 PXD056413。
DDA 和 DIA 分析
使用 MSFragger (v. 4.1) FragPipe (v. 22.0) 處理 DDA 數據,以鑒定肽和蛋白質,并生成用于 DIA 數據分析的光譜庫。根據人類 SwissProt 數據庫(2024 年 4 月 16 日從 UniProtKB 下載,僅限規范,40,870 個條目,包含 20,435 個反向誘餌)搜索 MS/MS 譜圖。將母離子和子離子的質量精度設置為 20 ppm,并啟用質量校準和參數優化。將酶特異性設置為胰蛋白酶/P,允許最多一個缺失的切割位點。半胱氨酸的氨基甲酰甲基化被設置為固定修飾,蛋氨酸的氧化和蛋白質 N 末端的乙酰化被允許為可變修飾。使用 Philosopher 中的 PeptideProphet 和 ProteinProphet 過濾結果,FDR < 1%。用于 DIA 數據分析的光譜庫是根據所有 DDA 數據的結果生成的。
對于 DIA 數據,通過 FragPipe (v. 22.0) 用 DIA-NN (v. 1.8.2 beta 8) 定量蛋白質,蛋白質水平 FDR < 1%。針對不同的運行分別確定質量精度和掃描窗口。定量結果使用 MetaboAnalyst (v. 6.0) 和 PlotsOfData 可視化。
自上而下的樣品制備、PEPPI 分級分離和處理
在手鑄的 10% SDS-PAGE 上分離 9 個 40 μg 的 HCPE 樣品,切除對應于 0-30 kDa 的區域,并如上所述進行 PEPPI。如上所述,用 MCW 、 FASP 和 SP3 方法處理 3 組。請注意,在濃縮之前,將 AnExSP 處理的樣品在液氮中快速冷凍。使用 SpeedVac 進行溶劑蒸發,不加熱,直到樣品體積減少至 ~5 μL(約 60–120 分鐘;因樣品而異)。濃縮后,使用 0.1% (v/v) FA、4.9% (v/v) 乙腈的 LC-MS 級水溶液將樣品體積定為 20 μL。
色譜和自上而下的質譜
通過納米毛細管高性能液相色譜在線與 EasySpray 納離子噴霧離子源(Thermo Fisher Scientific,加利福尼亞州圣何塞)聯用,進一步分離重懸的 HCPE 餾分(FASP 和 SP3 樣品進樣體積為 2 μL,MCW 進樣體積為 3.5 μL)。使用Ultimate 3000色譜系統(Thermo Fisher Scientific)進行反相液相色譜,將流動相B的梯度從5%提高到2 min從5%提高到14%,然后在50 min內從14%提高到42%,然后連續兩次以85% B清洗色譜柱,持續1 min,最后以5% B再平衡相,持續8.5 min, 保持 1.5 μL/min 的流速。流動相A由4.9% (v/v)乙腈水溶液在0.1% (v/v) FA中存在而成,而流動相B由4.9% (v/v)水的乙腈溶液和0.1% (v/v) FA組成。所有流動相組分均為LC-MS純度等級(Fisher Scientific)。將樣品直接進樣到帶有集成發射器的 MAbPac EasySpray 色譜柱上(Thermo Fisher Scientific;粒徑 4 μm,長度 15 cm,內徑 150 μm),使用集成的 EasySpray 柱溫箱加熱至 55 °C。通過施加 2.1–2.2 kV 電位產生納電噴霧。所有質譜測量均在配備 FAIMS Pro 裝置的 Orbitrap Eclipse 三組質譜儀 (Thermo Fisher Scientific) 上進行。在“蛋白質模式”下進行自上而下的實驗,離子路由多極壓力為 3 mTorr。源區域參數包括加熱傳輸毛細管的 320 °C 溫度、30% RF 振幅和 15 V 源內碎裂,以促進脫溶劑和去除不穩定的加合物。對于 DDA,使用 5 × 105充電和 50 ms 的自動增益控制 (AGC) 目標,最大進樣時間為 50 ms,最大進樣時間為 50 ms,在 400–2000 m/z 窗口內以 120,000 分辨能力(200 m/z 時)以 120,000 m/z 的分辨率(即單次微掃描)鑒定母離子。選擇四極桿(3 m/z 隔離窗口)進行 HCD MS2 碎裂(35% 歸一化碰撞能量),在 5–50+ 的電荷態范圍內,強度閾值為 2.5 × 104。在 Orbitrap 質量分析器中收集碎裂譜圖,在 400–2000 m/z 窗口內,無光譜平均,AGC 目標為 2.5 × 105 次充電,最大進樣時間為 500 ms。FAIMS 補償電壓 (CV) 在單次運行中變化,每個樣品單獨運行 3 次(每個樣品總共 9 個不同的 CV), 與之前的報告類似;(29) 運行 1:?40、?20 和 0 V;運行 2:?60、?50 和 ?30 V;運行 3:?10、5 和 15 V;每個 CV 使用 1.2 s 的循環時間。應用動態排除(持續時間 30 s)。所有質譜數據文件均已上傳到 MassIVE(存儲庫編號 MSV000095992)
自上而下的數據分析
將來自所有三種處理方法的 RAW 文件與 ProSight PD v. 4.2 (Proteinaceous, Inc., Evanston, IL, USA) 一起搜索,在 Proteome Discoverer 3.0 環境 (Thermo Fisher Scientific) 中作為節點運行,對照人類數據庫。使用提供的 High-High 處理和共識工作流程進行數據分析。對于 FAIMS 文件,Spectrum Selector (光譜選擇器) 節點中的 FAIMS CV 設置未指定,以適應每個文件使用多個 CV。所有譜圖都在 High-High cRAWler 節點內進行去卷積:使用 Xtract 利用滑動窗口算法從 MS1 譜圖中去除母離子質量數,掃描偏移量為 1,合并容差為 30 ppm,同時要求在至少 3 個滑動窗口檢測中檢測至少 3 種電荷態。在共識工作流程中定量之前,特征組需要 2.2 Da 的耐受性。應用了兩個數據庫搜索:使用 2.2 Da 母離子質量容差的注釋蛋白形式搜索,以及基于 15 ppm 母離子容差的子序列搜索。對于這兩個檢索,片段容差均設置為 10 ppm。由單個前體產生的蛋白形式數量限制為 1。從 ProSight PD 中鑒定出的蛋白質形式和 UniProt 登錄號以 1% FDR 過濾(對蛋白質形式、同工型和 UniProt 登錄水平進行三次單獨的 FDR 計算)。完成數據庫搜索后,針對每種處理方法過濾生成的 tdReport,并根據開發人員的建議重新計算 Q 值。對于水腫總平均值 (GRAVY) 和等電點 (pI) 分析,使用 GraphPad Prism 10 (GraphPad Software, Boston, MA, USA) 中的內置函數,使用 Kruskal-Wallis 和 Dunn 多重檢驗校正,根據每組的平均值進行統計檢驗。使用 DeepTMHMM 預測鑒定蛋白質的跨膜狀態。繪圖是使用 GraphPad Prism 10 生成的。
結果與討論
通過 SP3 純化 PEPPI 組分
我們嘗試根據先前發布的 BUP15 方案,以 250 μL 的樣品規模(典型的 PEPPI 餾分體積)建立 TDP 的 SP3 方案。使用的磁珠是兩種不同羧酸鹽修飾磁珠的等重混合物,即 Cytiva Sera-Mag SpeedBead 羧酸鹽修飾磁珠的親水和疏水版本。在 BUP 的 SP3 樣品制備中,通常建議在蛋白質中添加 10 倍重量的磁珠。 為了應用于 TDP,我們從 10 μg HCPE 中生成了四種不同 MW 范圍的 PEPPI 級分,并檢查了當添加不同量的珠子時與珠子結合的蛋白質量(圖 S1)。在 250 至 20 kDa 的 MW 范圍內,無論添加的磁珠量如何,回收率都相似,但在低于 20 kDa 的范圍內,當添加的磁珠量小于 500 μg 時,回收率略有下降。因此,我們在本研究中將珠子的量設置為 500 μg。
為了在室溫 (23–25 °C) 下快速回收磁珠上緊密吸附的蛋白質,我們認為需要表面活性劑輔助,并選擇使用 SDS,它可以被 AnExSP 去除。具體來說,我們將 20 μL 0.05% (w/v) SDS/100 mM ABC(之前已被證明適用于 AX-StageTip 純化)與洗滌的珠子混合,并在室溫下用試管振蕩器搖動混合物(圖 1A)。HCPE 分析表明,0.05% (w/v) SDS/100 mM ABC 可以在 10 分鐘內從珠子中回收蛋白質(圖 1B)。作為 SDS 的替代品,我們還研究了 RapiGest、尿素、NDSB-195 和辛基葡糖苷的使用,這些物質可以在 MS 分析前輕松去除。尿素、NDSB-195 和辛基葡萄糖苷在搖動 10 分鐘后沒有產生任何顯著的蛋白質回收(圖 S2),可酸降解表面活性劑 RapiGest 產生相當于 SDS,但隨后的酸處理導致回收的蛋白質降解(圖 S3)。因此,我們決定在本研究中繼續使用 SDS 進行蛋白質回收。
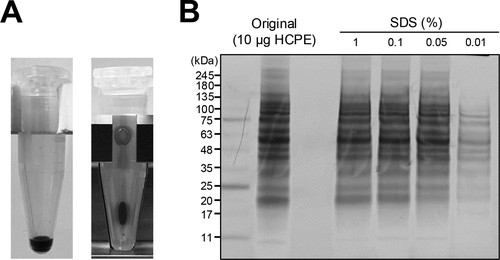
圖 1.使用 SDS 溶液從 SP3 微珠中回收蛋白質。(A) 從 SP3 微珠中回收蛋白質。通過在 23 °C 下在 20 μL 0.05% (w/v) SDS/100 mM ABC 中搖動珠子 10 分鐘(左圖),回收吸附在 SP3 珠子 (500 μg) 上的 HCPE (10 μg)。蛋白質回收后,將試管放在磁力架上以去除磁珠(右)。(B) 從不同 SDS 濃度 (0.01–1% (w/v)) 的磁珠中回收的蛋白質的 SDS-PAGE 圖像。條帶用 CBB 染色。SP3磁珠請參考 http://www.posuichina.com/Product/1736805216.html
PEPPI-SP3 工作流程
基于上述結果,我們建立了一個新的實驗工作流程 PEPPI-SP3,通過在 AnExSP 純化后結合基于 SP3 的蛋白質回收來純化 PEPPI 組分(補充方案)。PEPPI-SP3 工作流程的方案如圖 2 所示。在工作流程中,蛋白質組學樣品通過 SDS-PAGE 分離,然后用 PEPPI 進行分級分離。將乙醇添加到 500 μg SeraMag 珠子和所得 PEPPI 級分中,以產生 80% (v/v) 乙醇溶液,并劇烈搖動以將沉淀的蛋白質成分吸附到珠子上(圖 S4A 和 S4B)。使用磁力架分離珠子,并用 80% (v/v) 乙醇洗滌兩次,用乙腈洗滌一次,以去除 CBB 和 SDS(圖 S4C-S4E)。通過用 0.05% (w/v) SDS/100 mM ABC 搖動珠子 10 分鐘,從珠子中回收蛋白質,并在 AnExSP 純化后進行 LC-MS。
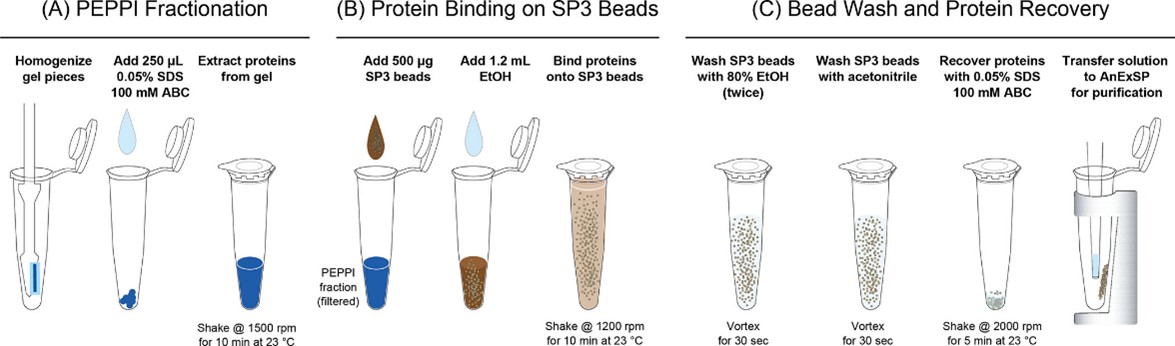
圖 2.用于自上而下的蛋白質組學的 PEPPI-SP3 工作流程。(A) PEPPI 分級分離。樣品 SDS-PAGE 分離后,用 CBB 對分離的蛋白質進行染色,并切除樣品泳道中所需的分子量范圍。使用一次性塑料搗碎管對切除的凝膠塊進行勻漿,并用 0.05% (w/v) SDS/100 mM ABC 被動提取凝膠中的蛋白質。(B) 蛋白與 SP3 微珠的結合。將蛋白質回收溶液(PEPPI 組分)與 SP3 珠子和乙醇混合,攪拌,沉淀的蛋白質吸附到珠子上。(C) 磁珠洗滌和蛋白質回收。用 80% (v/v) 乙醇洗滌珠子兩次,用乙腈洗滌一次,然后與 20 μL 0.05% (w/v) SDS/100 mM ABC 混合,以從珠子中回收蛋白質;攪拌 5 分鐘后,用磁鐵去除磁珠,并用 AX-StageTip 純化所得蛋白質溶液。SP3磁珠請參考 http://www.posuichina.com/Product/1736805216.html
將吸附在磁力架上的磁珠風干 10 分鐘。傳統的 PEPPI 工作流程使用 (1) PEPPI-MCW 與 MCW 沉淀或 (2) PEPPI-FASP 結合 FASP 和 AnExSP。PEPPI-MCW 從電泳到純化的時間為 2.3 小時,PEPPI-FASP 為 5.3 小時,而 PEPPI-SP3 需要 3.4 小時(圖 S5);FASP 是一個相對耗時的過程,通過使用 SP3 已得到極大的改進。
接下來,我們通過與 PEPPI-MCW 和 PEPPI-FASP 的比較驗證了 PEPPI-SP3 的性能。根據相應的工作流程處理來自 10 μg HCPE 的 PEPPI 級分,并使用納米 LC-MS 通過 DIA 進行 SDS-PAGE 和定量蛋白質組學。圖 3 顯示了從每個工作流程獲得的 PEPPI 級分的 SDS-PAGE 結果,CBB 染色圖像顯示,在 100 kDa 以上的高 MW 區域和低于 20 kDa 的低 MW 區域,工作流程之間存在很大差異。我們以前的研究已經表明,AnExSP 不適用于高 MW 蛋白;與 MCW 相比,使用 AnExSP 進行蛋白質純化(FASP 和 SP3)的工作流程對 100 kDa 以上的蛋白質的回收率往往較低。在低于 20 kDa 的 MW 區域,已知通常會導致低 MW 蛋白回收損失的 MCW 加工顯示,與 FASP 和 SP3 相比,回收的蛋白質量預期減少。最后,比較 FASP 和 SP3,注意到用 SP3 處理的餾分具有比 FASP 更高的譜帶密度,表明 SP3 在小于 20 kDa 的區域具有絕對優勢,這是當前 TDP 分析的主要目標區域。通過 ContamSpot 分析評估純化級分中 SDS 的水平,顯示 SP3 處理的級分的 SDS 水平低于 0.002% (w/v),并且純化結果與 MCW 相當(圖 S6)。即使將 HCPE 從 10 μg 增加到 40 μg(圖 S7),每個工作流程對餾分的純化結果與 10 μg 時的結果相似,SP3 在 20 kDa 以下的性能明顯好得多。
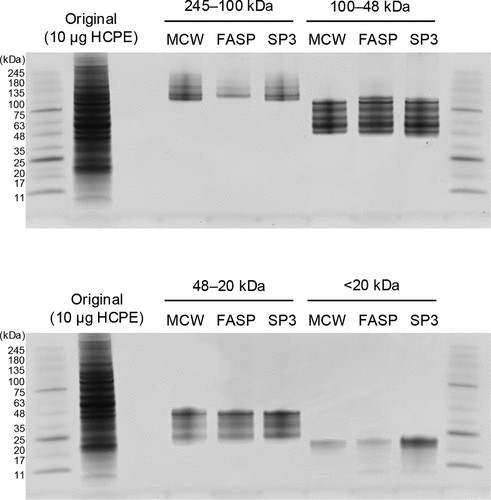
圖 3.通過三種不同方法純化的 PEPPI 組分的 SDS-PAGE 圖像。通過三種不同的方法(MCW、FASP和SP3)純化來自10 μg HCPE的四種PEPPI組分(245-100 kDa、100-48 kDa、48-20 kDa;<20 kDa),并通過SDS-PAGE再次分離這些組分。凝膠分離的組分用 CBB 染色。
對于 DIA 分析,將組分合并,用胰蛋白酶/Lys-C 消化,并進行全蛋白質組定量(圖 4A)。我們的分析在所有三個工作流程中共檢測到 5379 個蛋白質(表 S2),每個工作流程中檢測到的數量幾乎沒有差異(圖 4B),并且在所有三個工作流程中都檢測到許多組分 (5016)。圖 S8 顯示了兩種不同工作流程(SP3 與 MCW 或 SP3 與 FASP)之間 DIA 定量的比較結果:在 SP3 與 MCW 的情況下,每種方法顯示顯著變化的蛋白質數量(>3 倍,p < 0.05)相似(MCW:229 和 SP3:197)。在這些蛋白質中,在 MCW 中檢測到的蛋白質顯示更多氨基酸長度較長的蛋白質,而在 SP3 中檢測到的蛋白質顯示更多氨基酸長度較短的蛋白質(<300 個氨基酸)(圖 4C),這一趨勢與 SDS-PAGE 結果中觀察到的趨勢一致。在 SP3 與 FASP 的情況下(圖 S8),顯示顯著差異的蛋白質在 SP3 中的豐度是 FASP 中的三倍(FASP:76 和 SP3:220),并且 SP3 中豐富的大多數蛋白質是氨基酸殘基少于 300 個的蛋白質(圖 4C)。這些結果表明,當目標蛋白質是低 MW 蛋白時,SP3 是首選工作流程。事實上,當考慮組蛋白(TDP 中蛋白質形式分析的重要靶標)時,SP3 對本研究中檢測到的所有 15 種組蛋白都顯示出比其他兩種更好的恢復結果(圖 S9)。相比之下,對于 TDP 分析仍未開發的高 MW 蛋白,MCW 將是首選工作流程。我們還使用 DIA 數據評估了每個工作流程的定量準確性。圖 4D 顯示了小提琴圖中每個工作流程的變異系數 (CV) 分布,表明 SP3 在定量準確度方面優于其他工作流程。與其他工作流程相比,FASP 的定量精度較低,這可能是由于超濾過濾器上的蛋白質吸附損失所致。
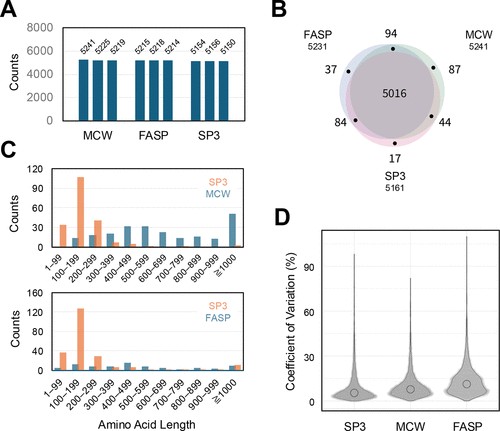
圖 4.通過 DIA 定量評估不同的 PEPPI 工作流程。(A) 使用 PEPPI-MCW (MCW)、PEPPI-FASP (FASP) 和 PEPPI-SP3 (SP3) 三種 PEPPI 工作流程從 10 μg HCPE 中鑒定的蛋白質的比較,混合賴斯-C/胰蛋白酶,并通過 LC-MS 進行 DIA 分析。(B) 維恩圖顯示了每個工作流程中鑒定的蛋白質之間的關系。(C) 蛋白質中氨基酸長度的比較,如圖 S8 所示的顯著差異。(D) 每個工作流程中鑒定的蛋白質組的 CV 分布。圓圈表示中位數。
PEPPI-SP3 組分的 TDP 分析
接下來,我們通過將 PEPPI-SP3 方法與 PEPPI-MCW 和 PEPPI-FASP 方案進行比較,驗證了 PEPPI-SP3 方法對 TDP 的性能。為了進行此比較,對 40 μg HCPE 的三個技術重復進行 PEPPI 分級,以生成每種處理方法的 0-30 kDa 餾分(圖 S10)。在用相應的方法(MCW、FASP 和 SP3)處理后,使用 FAIMS-HiHi 方法分析樣品,該方法依賴于內部補償電壓 (CV) 步進,(32) 三次運行中的每一次都使用三個不同的 CV 值(即,每個樣品總共經受 9 個 CV)以增加所研究蛋白質組的深度。
總體而言,PEPPI-SP3 方法在多個指標上表現出優于其他兩種方法的顯著優勢。相對于 MCW 方法,它顯著增加了 60% 的 UniProt 種質數量和 85% 的蛋白質組鑒定數量(圖 5A)。雖然與 MCW 方法相比,FASP 方法確實增加了鑒定結果,但它比 SP3 的鑒定數量少了約 20%。根據對 ?40 V 補償電壓的總離子色譜圖 (TIC) 的目視檢查,鑒定數量的差異并不奇怪,該 CV 值導致每種方法鑒定出大多數蛋白質形式(圖 S11)。通過將全局 TIC 強度歸一化為 SP3 樣品,FASP 樣品顯示信號降低 ~ 35%。然而,當在 SP3 和 MCW 之間進行類似的比較時,盡管 MCW 樣品的負載量高出 75%,但 MCW 的最大強度降低了 ~50%。這表明與 FASP 和 SP3 方法相比,MCW 的蛋白質組回收率較低。此外,SP3 處理樣品的復雜性(根據色譜圖中存在的不同洗脫峰的數量估計)高于其他兩種處理方法中的任何一種。
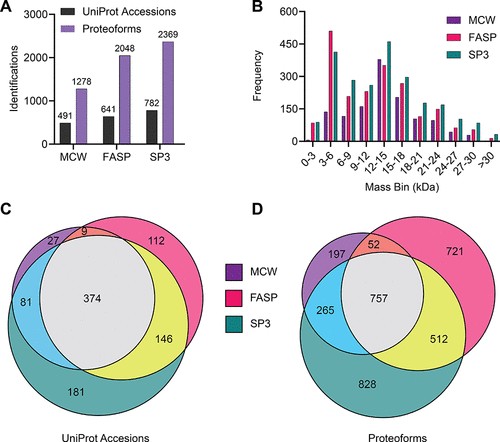
圖 5.三種治療方法的比較總結。(A) 已鑒定的 UniProt 登錄號和獨特蛋白形式的全局計數。(B) 已鑒定的蛋白質形式的質量分布。(C) 已識別的 UniProt 登錄號的維恩圖。(D) 已鑒定的蛋白質形式的維恩圖。三種治療方法的結果根據圖中的圖例進行顏色編碼
我們對三種治療方法之間主要差異的調查揭示了 FASP 和 SP3 方法的獨特優勢。一般來說,SP3 方法在整個 0–30 kDa 質量范圍內優于 MCW 和 FASP 方法(圖 5B)。唯一值得注意的例外是 3–6 kDa 質量 bin,其中 FASP 方案鑒定出的蛋白質形式多出 25%。FASP 和 SP3 方法對低于 9 kDa 的蛋白質形式具有顯著優勢,導致在該范圍內鑒定的蛋白質形式比 MCW 處理的樣品多 >200%。在聚合 0-9 kDa 范圍內,FASP 方案鑒定的蛋白質形式略多于 SP3 方案(分別為 803 和 785)。然而,在較高分子量范圍 (>15 kDa) 下,SP3 方法與 MCW 方案相比保持了相當大的優勢(鑒定結果提高了約 80%),而以前在研究人血清蛋白質形式時,MCW 方法的性能與 FASP 方案相當。在相同的 15–30 kDa 質量范圍內,SP3 樣品中鑒定的蛋白質形式比 FASP 樣品多 30%(表 S3–S5)。
雖然 PEPPI-SP3 方法在 0-30 kDa 范圍內實現了更多的鑒定,但我們想確定三種方法鑒定之間的重疊程度。在查看鑒定出的 UniProt 種質數量時,SP3 方法鑒定出 MCW 方法鑒定的 93% 和 FASP 方法鑒定的 81%(圖 5C)。然而,當我們進入蛋白質組水平時,我們觀察到這三種方法之間具有更高程度的唯一性。在 MCW 和 FASP 樣品中鑒定出的與 SP3 相同的蛋白質形式的百分比分別降低到 80% 和 62%(圖 5D)。
接下來,我們檢查了質譜分析的技術變異性。我們首先比較了給定處理方法的技術重復的色譜圖。在檢查各種 CV 時,目視檢查沒有顯示技術重復之間的實質性差異(圖 S12A 中可以找到 SP3 技術重復的 CV – 40 V 示例)。由于色譜圖的差異很小,我們研究了每種處理方法的 MS 運行之間蛋白質組鑒定的重疊情況(圖 S12B)。在比較每次 MS 運行中每種處理方法的所有三個技術重復中鑒定出的蛋白形式數量時,我們觀察到 MCW 技術重復在其三個技術重復中共享的已鑒定蛋白質形式的百分比略高于 FASP 或 SP3 技術重復。每次 MS 運行都觀察到此趨勢。我們將技術變異性至少部分歸因于 DDA 中前體選擇的隨機性,以及用 FASP 和 SP3 方法處理的樣品復雜性增加(即,較大的蛋白質形態異質性,較低的鑒定重現性)。可以使用多種措施來減輕這一限制,其中包括專門增加基于分子量的 PEPPI 餾分的數量以進行分析和/或使用更長的色譜梯度。使用 SP3 方法處理的單個樣品的 TIC 痕量具有高度重現性,并且與 MCW 樣品相比,在所有重復中鑒定的蛋白質形式分數僅略有降低,這表明 SP3 方法可能適用于基于無標記蛋白質形式定量的定量 TDP 研究。
雖然質譜水平的技術差異可以解釋高度唯一性的一些原因,但它不太可能是唯一的原因。下一步調查是確定各種治療方法中是否存在支持或反對特定翻譯后修飾的偏倚。由于三種方法之間鑒定的蛋白質形式數量存在很大差異,因此我們將給定 PTM 的頻率標準化為鑒定出的蛋白質形式數量,以產生每 100 個識別蛋白質形式的比率。然后,我們根據兩種方法之間比率的 Log2 轉換對 10 個最常見識別的 PTM 進行了成對比較(圖 S13)。綜上所述,FASP 方法比 MCW 方法更能識別截斷產物,但不如 SP3 方法。同時,與 MCW 和 SP3 方法相比,FASP 方法在識別蛋白質 N 端和賴氨酸的乙酰化事件以及絲氨酸、蘇氨酸和酪氨酸殘基的磷酸化事件方面表現出乎意料地差。即使僅考慮 C 評分為 ≥40 的蛋白質形式的 PTM 分析,也可以獲得類似的結果,這些蛋白質形式是表征最好的蛋白質。這些觀察結果表明,雖然 FASP 擅長識別小的蛋白質形式(即截斷產物),但它難以保留對信號轉導很重要的 PTM。由于 FASP 和 SP3 方法之間的差異發生在共享使用 AX-StageTip 之前,因此可以推測 FASP 中使用的離心濃縮器膜正在吸附其中一些蛋白質形式。在 PTM 鑒定方面,SP3 方法對于十種最常見的修飾,尤其是截斷產物,其性能與 MCW 方法一樣好或更好。通過分析所有三個數據集之間共享并攜帶特定 PTM 的蛋白質形式的前體離子強度,證實了這一點。結果表明,與 MCW 和 SP3 樣品相比,使用 FASP 清潔的蛋白質形式的信號(特別是攜帶乙酰化和磷酸化)相對較弱,相反,它們顯示出相當的信號強度值(圖 S14)。
基于這些結果,我們試圖確定其他物理化學性質是否區分了應用三種測試處理方法后鑒定的蛋白質形式。雖然通過 GRAVY 分析計算的疏水性程度在三個蛋白群之間沒有顯著差異,但三個組之間的等電點分布在統計學上不同 (p < 0.0001)(圖 S15)。使用 MCW 方法鑒定的蛋白質形式的特點是中位數和平均 pI 分別為 9.7 和 8.9,這與使用 SP3 方法鑒定的蛋白質形式的相應值(中位數和平均 pI 分別為 9.1 和 8.4)相差不遠;然而,SP3 方法也導致鑒定出更多的酸性蛋白形式 (第一個四分位數的 pI:MCW 和 SP3 分別為 7.0 和 6.4)。相比之下,FASP 方法的 pI 分布包括較少的基本蛋白質形式 (中位數和平均值分別為 7.0 和 7.3)。如圖 S15B 所示,SP3 方法的 pI 分布介于其他兩種方法之間,這表明這種方法能夠幾乎與 MCW 方法一樣回收堿性蛋白質形式,同時還保留了大部分由 FASP 方法富集的中性和酸性蛋白質形式。
最后,使用 DeepTMHMM 進行的蛋白質跨膜分析表明,SP3 處理有利于恢復和鑒定預測為跨膜的蛋白質形式(占該方案獲得的鑒定總數的 6.5%),優于 MCW(占總數的 5.5%),特別是 FASP(僅占總數的 2%)方法(圖 6)。
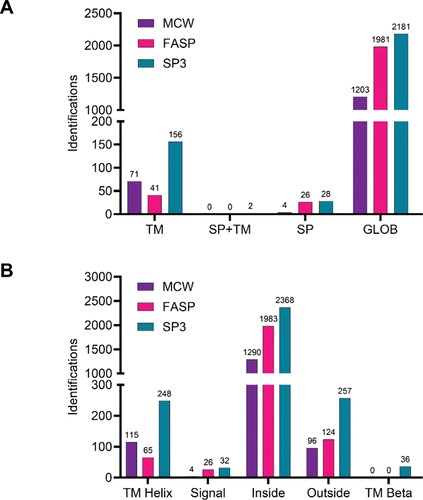
圖 6.使用 DeepTMHMM 預測蛋白質形態拓撲。(A) 使用 MCW、FASP 和 SP3 去除去污劑后從樣品中鑒定的蛋白質形式的拓撲類型。TM:跨膜,SP+TM:信號肽的跨膜,SP:信號肽,GLOB:球狀。(B) 在 proteoforms 中鑒定的預測結構域。TM 螺旋 = α 螺旋跨膜,信號:信號肽,內部:細胞內/胞質溶質,外:ER/高爾基體/溶酶體的細胞/腔外,以及 TM Beta:β桶跨膜。一旦對 ID 總數進行標準化,FASP 處理后跨膜蛋白形式就會減少。
結論
在第一個 PEPPI 工作流程中,已經被廣泛用作 TDP 的樣品預分餾方法,純化介質(通常會導致低 MW 蛋白損失)或 FASP(耗時)已被用于純化 PEPPI 組分。在這項研究中,我們開發了 PEPPI-SP3,這是最新的和第三個 PEPPI 工作流程,它使用 SP3 微珠進行了穩定而簡單的蛋白質純化。建立回收吸附在 SP3 磁珠上的完整蛋白質的工藝對于實現 PEPPI-SP3 至關重要,這是通過將室溫下快速回收與 0.05% (w/v) SDS 和使用 AX-StageTip 去除 SDS 相結合來實現的。AnExSP 將餾分成功純化至與 MCW 相當的水平,并且通過 LC-MS 測量 TDP 時未觀察到問題。PEPPI-SP3 在回收低于 20 kDa 的低分子量范圍內的蛋白質方面優于傳統方法,非常適合 TDP,并且由于 SP3 支持磁珠上加工,PEPPI-SP3 還可以增強 BUP 和中下蛋白質組學中的樣品預處理。 盡管 SP3 在 BUP 中被確立為一種通用的樣品制備方法,但它在 SDS-PAGE 后應用于凝膠中的蛋白質仍然難以捉摸。本研究中建立的 SDS-PAGE 到 SP3 通路最終允許在基于凝膠的自上而下分析中使用 SP3。
隨著 PEPPI-SP3 在本研究中的成功開發,現在有三種有效的方法可用于純化 PEPPI 組分(MCW、FASP 和 SP3),但為 TDP 選擇合適的工作流程對于獲得最佳結果至關重要。在作時間方面,SP3 純化可以在比 FASP 更短的時間內完成,盡管它仍然比 MCW 慢。在可作性方面,當蛋白質量較少時,MCW 在使用中具有挑戰性,因為通常很難目視確認形成的蛋白質沉淀,而且 FASP 易于執行但耗時,而 SP3 具有無論樣品量如何都穩健和簡單的理想組合。本研究中通過定量 DIA 分析進行的比較評估也支持 SP3 在定量準確性方面的優越性,使 SP3 成為在 TDP 中純化 PEPPI 組分的首選工作流程,尤其是在與低 MW 蛋白(如組蛋白)的相容性方面。對于低于 30 kDa 的蛋白質形式的 TDP 分析,與 MCW 和 FASP 相比,SP3 方法提供了最多的鑒定數量,同時還能夠保留與信號轉導通路相關的 PTM。
盡管用當前的 TDP 很難分析超過 100 kDa 的高 MW 蛋白,但仍然可以應用 PEPPI,但對如此高 MW 的蛋白,MCW 是比 SP3 更好的選擇。MCW 也可能具有成本優勢,因為它不需要 SP3 微珠或超濾設備。PEPPI 的性質允許在樣本數量較多時使用兩種不同的方法。例如,可以在各自的 MW 范圍內同時使用 SP3 和 MCW 以降低成本。但是,SP3 仍有降低成本的空間。近年來,SP4 方案通過離心而不是磁力架來回收磁珠,已被報道為 SP3 的改進方案,允許用低成本的非磁珠替換磁珠。基于低成本協議的改進將使潛在的 PEPPI-SP4 更加便宜。
TDP 的臨床應用在不久的將來可能會加速,對大量樣本的高通量處理的需求將相應增加。隨著樣品數量的增加,MCW 和 FASP 的繁瑣作可能是一個主要障礙。相比之下,市售的自動化設備可用于 SP3,并且在 BUP 的多樣品處理中表現出優異的性能。BUP 中 SP3 自動加工的關鍵技術應可轉移到 TDP 的 PEPPI 分餾加工。SP3磁珠請參考 http://www.posuichina.com/Product/1736805216.html
《使用 SP3 純化基于凝膠的樣品分級用于自上而下的蛋白質組學》總結
研究背景
1. 蛋白質組學研究目標:了解龐大的人類蛋白質組,需要開發全面的蛋白質組分析技術。
2. 自上而下蛋白質組學(TDP)的需求:自下而上蛋白質組學難以準確鑒定蛋白質形式,TDP 可直接分析完整蛋白質形式,但需樣品預分餾以檢測更多蛋白質組分。
3. 現有方法的問題:SDS - PAGE 是常用預分餾方法,結合 PEPPI - MS 可高效提取凝膠中蛋白質,但獲得的餾分含干擾 MS 分析的 CBB 和 SDS,當前蛋白質純化方法(如 MCW、AnExSP)存在低分子量蛋白損失、操作繁瑣等問題。
實驗方法
1. 材料:使用 MS 相容的人蛋白提取物,經還原、烷基化處理,調整蛋白質濃度。
2. SDS - PAGE:使用預制凝膠或自制凝膠,不同染色劑染色。
3. PEPPI 分級分離:切取染色凝膠感興趣 MW 區域,研磨凝膠塊,用含 SDS 的碳酸氫銨溶液提取蛋白質,得到 PEPPI 組分。
4. 蛋白質純化
MCW:加入甲醇、氯仿和水,離心分層,去除上層,洗滌下層蛋白質沉淀。
FASP:通過超濾裝置置換溶劑,用 AX - StageTip 純化。
SP3:將 PEPPI 組分與 SP3 珠子和乙醇混合,吸附蛋白質,洗滌珠子,用含 SDS 的碳酸氫銨溶液回收蛋白質,再用 AX - StageTip 純化。
5. 檢測與分析
ContamSpot 檢測:測定純化后 PEPPI 組分的殘留 SDS 濃度。
胰蛋白酶 / Lys - C 消化:溶解 PEPPI 組分,用胰蛋白酶 / 賴氨酸 - C 混合物消解。
NanoLC/MS/MS:獲取 MS 和 MS/MS 數據,進行 DDA 和 DIA 分析。
自上而下的樣品制備與分析:對 HCPE 樣品進行 PEPPI 分級,用不同方法處理,通過色譜和質譜進一步分離和分析,使用 ProSight PD 等軟件進行數據分析。
結果與討論
1. 通過 SP3 純化 PEPPI 組分
確定磁珠添加量:在 250 至 20 kDa MW 范圍,磁珠添加量對回收率影響小;低于 20 kDa 時,磁珠量小于 500 μg 回收率略降,因此設定磁珠量為 500 μg。
選擇蛋白質回收試劑:研究多種試劑,發現 0.05%(w/v)SDS/100 mM ABC 可在 10 分鐘內從珠子中回收蛋白質,其他試劑效果不佳,最終選擇 SDS 用于蛋白質回收。
2. PEPPI - SP3 工作流程
建立新流程:結合基于 SP3 的蛋白質回收和 AnExSP 純化,建立 PEPPI - SP3 工作流程。該流程中,蛋白質組樣品經 SDS - PAGE 分離、PEPPI 分級,與 SP3 珠子結合,洗滌后回收蛋白質,再經 AnExSP 純化進行 LC - MS 分析。
流程時間比較:PEPPI - MCW 時間為 2.3 小時,PEPPI - FASP 為 5.3 小時,PEPPI - SP3 為 3.4 小時,SP3 改進了 FASP 耗時的問題。
性能驗證:SDS - PAGE 結果顯示,在高 MW 區域(>100 kDa),使用 AnExSP 的工作流程(FASP 和 SP3)對蛋白質回收率低于 MCW;在低 MW 區域(<20 kDa),MCW 加工導致低 MW 蛋白回收損失,SP3 處理的餾分譜帶密度高于 FASP。ContamSpot 分析表明 SP3 處理的餾分 SDS 水平低于 0.002%(w/v),與 MCW 相當。DIA 分析顯示,三個工作流程共檢測到 5379 個蛋白質,SP3 在低 MW 蛋白回收方面表現更好,對組蛋白的恢復結果優于其他兩種方法,且在定量準確度方面優于其他工作流程。
3. PEPPI - SP3 組分的 TDP 分析
鑒定數量優勢:PEPPI - SP3 方法相對于 MCW 方法,顯著增加了 60% 的 UniProt 種質數量和 85% 的蛋白質組鑒定數量;與 FASP 方法相比,鑒定數量多約 20%。
質量范圍優勢:SP3 方法在整個 0 - 30 kDa 質量范圍內表現出色,在 > 15 kDa 質量范圍優勢明顯;FASP 方法在 3 - 6 kDa 質量 bin 鑒定出的蛋白質形式多出 25%,在 0 - 9 kDa 范圍鑒定的蛋白質形式略多于 SP3。
鑒定重疊情況:SP3 方法鑒定出 MCW 方法鑒定的 93% 和 FASP 方法鑒定的 81% 的 UniProt 種質;在蛋白質組水平,MCW 和 FASP 樣品中鑒定出的與 SP3 相同的蛋白質形式的百分比分別為 80% 和 62%。
技術變異性:SP3 方法處理的單個樣品 TIC 痕量重現性高,適用于定量 TDP 研究。
翻譯后修飾(PTM)鑒定:FASP 方法在識別某些 PTM(如蛋白質 N 端和賴氨酸的乙酰化、絲氨酸等殘基的磷酸化)方面表現較差,SP3 方法在 PTM 鑒定方面性能與 MCW 方法相當或更好。
蛋白質物理化學性質:SP3 方法鑒定的蛋白質形式等電點分布介于 MCW 和 FASP 之間,能回收堿性蛋白質形式,同時保留中性和酸性蛋白質形式;SP3 處理有利于恢復和鑒定預測為跨膜的蛋白質形式。
結論
1. 開發新工作流程:開發了 PEPPI - SP3 工作流程,通過室溫下用 0.05%(w/v)SDS 快速回收吸附在 SP3 磁珠上的完整蛋白質,并結合 AX - StageTip 去除 SDS,實現穩定而簡單的蛋白質純化。
2. 方法優勢:PEPPI - SP3 在回收低分子量(<20 kDa)蛋白質方面優于傳統方法,適合 TDP,也可增強 BUP 和中下蛋白質組學中的樣品預處理。
3. 工作流程選擇:為 TDP 選擇合適工作流程很關鍵,SP3 在定量準確性、操作穩健性和對低 MW 蛋白的相容性方面表現優越,是純化 PEPPI 組分的首選工作流程;對于高 MW 蛋白(>100 kDa),MCW 是更好選擇。
4. 未來展望:隨著 TDP 臨床應用需求增加,SP3 可借助市售自動化設備實現高通量處理,未來可通過改進降低成本,如采用 SP4 方案等。
- 上一篇:生物交聯技術——交聯劑功能及使用特點 2025/4/22
- 下一篇:SP3 和 SP4 捕獲蛋白質的差異展現了蛋白質組學純化技術 2025/4/15