

臨床糖蛋白質(zhì)組學研究方法綜述
2 臨床糖蛋白質(zhì)組學方法概述
2.1 臨床糖蛋白質(zhì)組學方法的基礎原理和邏輯基礎
糖基化后,產(chǎn)生功能性糖蛋白,在生物體系中發(fā)揮著關鍵作用。然而,這種修飾過程有時可能導致特定糖蛋白的異常產(chǎn)生,這些糖蛋白的糖位點和/或糖鏈以病理方式異常改變,從而導致一系列功能變化。糖蛋白質(zhì)組學的方法涉及幾個關鍵步驟:臨床樣本選擇、樣本處理(蛋白質(zhì)提取、酶消化、IGP富集)、LC–MS/MS分析、生物信息學分析和結(jié)果驗證(圖2)。糖蛋白質(zhì)組學分析的第一個關鍵步驟是確保臨床樣本的質(zhì)量,因為它直接影響最終結(jié)果。復雜性更高、異質(zhì)性和糖蛋白濃度較低的臨床樣本在分析上提出了重大挑戰(zhàn)。在樣本處理之后,隔離和/或富集糖蛋白和/或IGP是關鍵步驟,因為糖基化的豐度有限。IGP富集的偏好可能存在于不同的富集材料或策略中。這種IGP富集過程通常與LC–MS/MS技術結(jié)合使用,已成為糖蛋白質(zhì)組研究中深入識別IGP的重要工具。選擇質(zhì)譜儀和碎裂模式是至關重要的,因為肽鏈和糖鏈碎裂需要不同的碎裂能量。這個選擇直接影響IGP識別的準確性和/或深度。注釋IGP MS/MS譜圖是一個具有挑戰(zhàn)性的任務,涉及正確分配肽載體、糖位點和附著的糖鏈。已經(jīng)開發(fā)了許多軟件程序和算法(Byonic、MSFraggerGlyco、pGlyco系列、StrucGP等),用于糖蛋白質(zhì)組學的識別和定量分析。盡管糖蛋白質(zhì)組學存在生物信息學障礙,但一些工具已經(jīng)顯示出卓越的性能和巨大潛力,提供了更敏感、準確和全面的糖基化信息。此外,基于LC–MS/MS的糖蛋白質(zhì)組學發(fā)現(xiàn)應通過額外的方法進行驗證,如小規(guī)模生化實驗和大規(guī)模隊列驗證。至關重要的是,應通過細胞和動物實驗確認糖蛋白的功能作用。總之,糖蛋白質(zhì)組學的復雜性強調(diào)了解碼糖蛋白及其生物重要性的謎題所需的復雜性和準確性。
2.2 臨床樣本選擇
在進行臨床糖蛋白質(zhì)組學之前,必須仔細考慮研究的每個方面,包括實驗設計、臨床樣本、設備和軟件。值得注意的是,臨床樣本(人體組織和體液)的選擇顯著影響實驗結(jié)果的可信度(圖2A)。特別是,應考慮以下方面:疾病亞型和階段、臨床數(shù)據(jù)、樣本類型、樣本分類、樣本量、收集程序、預處理方法、存儲條件、運輸物流等。例如,Cao等人對胰腺導管腺癌(PDAC)和正常鄰近組織的胰腺組織樣本進行了糖蛋白質(zhì)組學分析。他們收集了臨床數(shù)據(jù),并考慮了不同的組織來源和國家。對組織樣本的收集、預處理、存儲和運輸應用了標準化。在臨床糖蛋白質(zhì)組學中,通過穿刺和手術獲得的生理和病理組織樣本,以及通過臨床檢查獲得的體液(如血液、尿液、精液、眼淚、唾液和CSF)是最常見的來源(圖2A)。因此,實驗設計應包括適當?shù)膶φ战M和實驗組,確保每個組中的病例數(shù)量足夠(通常超過30個),樣本量足夠(通常確保至少提取100 μg蛋白質(zhì)),并最小化不必要的污染。此外,建立標準化的樣本收集、預處理、存儲和運輸協(xié)議對于有效進行后續(xù)實驗至關重要。必須嚴格遵守這些程序,以確保結(jié)果的可靠性和可重復性。
2.3 樣本處理
2.3.1 蛋白質(zhì)提取
臨床樣本處理是糖蛋白質(zhì)組學分析的一個重要瓶頸。為了減少實驗中的干擾,建立一個針對實驗目標和樣本類型的標準化流程至關重要。臨床樣本處理的第一步是蛋白質(zhì)提取(圖2B)。根據(jù)實驗目的,必須決定是提取所有蛋白質(zhì)還是針對特定的糖蛋白(如高豐度的免疫球蛋白[Ig]和尿調(diào)節(jié)蛋白)進行提取。為了有效權衡各種提取策略的優(yōu)缺點,我們提出了一個全面的方法。首先,人們應該仔細優(yōu)化和比較幾種糖蛋白質(zhì)組學提取技術,重點關注提取試劑和所采用的方法。隨后,選擇在大規(guī)模臨床樣本中提取糖蛋白質(zhì)組最有效的方法。最后階段,基于對特定感興趣糖蛋白的識別,可以針對這些目標糖蛋白定制提取過程。這種策略確保了在提取過程中平衡效率和特異性,有助于更精確和深入地分析糖蛋白質(zhì)組。此外,提取溶劑和方法的選擇應與樣本類型相匹配。例如,在處理最常用的臨床樣本人類血漿時,去除高豐度蛋白(HAPs)很重要。HAPs占人類血漿總蛋白質(zhì)組的85%,血漿蛋白的濃度跨越了超過10個數(shù)量級的動態(tài)范圍。這些HAPs可能導致質(zhì)譜信號抑制,限制了低豐度蛋白(LAPs)的檢測。已經(jīng)開發(fā)了許多方法來簡化和標準化血漿樣本。免疫基礎的耗竭技術,如針對最豐富的兩種或十四種蛋白質(zhì),以及組合肽配體庫的使用,在蛋白質(zhì)組學研究中常用。
近幾十年來,基于納米技術樣本處理策略已成為進行自動化血漿處理和深入蛋白質(zhì)組分析的高效方法。磁性納米粒子(MNs)體積小,比表面積大,親和力位點豐富,允許特異性或非特異性富集蛋白質(zhì)或PTMs(質(zhì)譜前處理試劑盒_生物磁珠專家)。當MNs進入血漿時,最初吸附HAPs在納米-血漿界面形成蛋白質(zhì)冠。這些HAPs隨后被具有更高親和力的LAPs所取代。尿液樣本以其臨床收集的便利性、無創(chuàng)性以及能夠提供大量和可持續(xù)的數(shù)量而聞名。然而,尿液蛋白質(zhì)組受到變異性和稀釋的影響,需要在數(shù)據(jù)分析中仔細考慮標準化程序。許多研究表明,尿液可以作為一系列疾病早期生物標志物的寶貴來源。組織樣本通過手術或穿刺獲得,是探索疾病進展分子機制的重要資源。蛋白質(zhì)提取過程中的均質(zhì)化非常重要,因為任何不完全的均質(zhì)化都可能導致關鍵信息的丟失,從而影響發(fā)現(xiàn)的準確性和精確性。組織樣本提取的常用方法包括研磨、均質(zhì)化和超聲處理等。基本原理圍繞最小化蛋白質(zhì)降解,防止污染,并在低溫和裂解液保存的受控環(huán)境中進行。
除了分析復雜臨床樣本的蛋白質(zhì)組外,通過抗體親和力和凝集素親和力對目標糖蛋白進行深入糖基化分析,對識別疾病生物標志物至關重要。例如,我們通過固定化蛋白A/G瓊脂糖從人血漿中分離出IgG,并檢查其在各種慢性腎臟疾病(CKDs)中的N-糖基化。此外,我們使用硅藻土粉末從人尿液中提取尿調(diào)節(jié)蛋白,并分析其在IgA腎病(IgAN)中的N-糖基化。總之,在選擇蛋白質(zhì)提取方法時,重要的是要考慮實驗目的和樣本類型,并遵循上述概述的基本原則。
2.3.2 酶消化
對于復雜臨床樣本中IGP的分析,通常不需要特殊的酶消化要求。通過自下而上的射擊蛋白質(zhì)組學(Figure 2B)進行IGP分析時,胰蛋白酶是主要使用的酶。然而,當分析特定的糖蛋白時,需要進行糖位點預測,以選擇適當?shù)拿富蛎傅慕M合。例如,重組的嚴重急性呼吸綜合征冠狀病毒-2(SARS-CoV-2)刺突蛋白含有22個潛在的N-糖基化位點。對酶切位點的理論分析表明,僅使用胰蛋白酶不能產(chǎn)生足夠長的肽段以覆蓋所有潛在的N-糖基化位點。為了解決這個問題,引入了內(nèi)蛋白酶Glu-C來識別缺失的N-糖基化位點。
同樣,Watanabe等人分別用胰蛋白酶、糜蛋白酶或α-溶細胞蛋白酶消化刺突蛋白,以覆蓋所有潛在的N-糖基化位點。肽N-糖苷酶F(PNGase F)是一種來自Elizabethkingia miricola的酶,通過大腸桿菌重組技術生產(chǎn)。它可以通過裂解幾乎所有類型的N-糖鏈(包括高甘露糖型、混合型和復雜型糖鏈)連接處的內(nèi)層N-乙酰葡萄糖胺(GlcNAc)和天冬酰胺殘基,有效地移除N-糖鏈。因此,PNGase F通常用于識別N-糖鏈和N-糖基化位點。此外,當需要O-糖基化分析時,使用PNGase F消除N-糖鏈,以最小化對O-糖鏈檢測的干擾。缺乏明確的蛋白質(zhì)序列一致性對于O-糖基化位點的識別是一個挑戰(zhàn)。然而,最近發(fā)現(xiàn)一種多功能的O-糖蛋白酶,稱為免疫調(diào)節(jié)金屬蛋白酶來自銅綠假單胞菌。這種酶可以特異性識別并切割O-糖基化絲氨酸或蘇氨酸殘基附近的糖蛋白,有助于準確識別每個O-糖基化位點的O-糖鏈結(jié)構。利用這種創(chuàng)新酶,研究人員在小鼠大腦中識別了近100個O-糖蛋白。除了上述特定的蛋白酶外,非特異性蛋白酶也可用于檢測特定的糖基化。例如,使用非特異性絲氨酸蛋白酶蛋白酶K對整體蛋白質(zhì)消化很有價值。使用各種酶及其組合將增強糖位點和糖鏈的全面識別。
2.3.3 IGP富集
在臨床樣本中,糖蛋白和IGP的豐度低,加上MS分析期間原生IGP的離子抑制和復雜的位點特異性異質(zhì)性,為準確檢測帶來了挑戰(zhàn)。為了解決這些挑戰(zhàn),已經(jīng)開發(fā)了各種富集方法來提高MS分析期間IGP的可檢測性和靈敏度。這些方法包括使用特定凝集素進行捕獲、基于水合硼酸的捕獲技術和親和分離策略。最常用的糖蛋白或IGP富集技術包括凝集素親和色譜法(LAC)、水合硼酸化學、親水作用液相色譜(HILIC)和水合肼化學。這些方法已經(jīng)迅速發(fā)展,以提高糖蛋白或IGP的富集效率。表1概述了每種富集方法的優(yōu)點和缺點。LAC已廣泛用于研究具有特殊糖鏈的糖蛋白。凝集素源自植物或動物,具有獨特的結(jié)合位點,能夠識別特定的糖鏈。這一特性使它們成為科學界幾十年來寶貴的工具,特別是在識別新的疾病相關生物標志物方面。水合硼酸化學方法是以其與具有順式1,2和1,3二醇的糖鏈的可逆反應而聞名,在堿性條件下形成環(huán)酯,在酸性條件下釋放糖鏈,同時保持其結(jié)構。HILIC方法作為一種新興的分離和富集方法,在糖蛋白質(zhì)組學中表現(xiàn)出色,因為它擅長分離IGP和糖鏈。這種技術利用離子、親水性IGP和相對疏水的非糖基化肽之間的對比電性質(zhì)。這種獨特的配置使HILIC能夠有效地在色譜柱上結(jié)合IGP,促進非糖基化肽的去除。水合肼化學方法用于氧化糖鏈通過水合肼試劑的修飾。它包括氧化、鏈接形成、蛋白水解、同位素標記、釋放和隨后分析等一系列步驟。這種高效的方法是探索糖位點的強大手段。
功能化的MNP方法已被用于富集糖蛋白,擴大了磁性納米材料的應用范圍。各種納米粒子作為膠囊綁定糖蛋白或IGP。例如,一種名為Fe3O4@mSiO2@G6P的親水性納米材料被特別設計用于從辣根過氧化物酶和IgG消化物中捕獲IGP。此外,一種超親水的介孔二氧化硅磁性納米球體,稱為Fe3O4–CG@mSiO2,以其出色的吸附能力、靈敏度、尺寸排除功能、穩(wěn)定性和回收效率而表現(xiàn)出色。這使得它在提取血清外泌體方面非常有效。因此,根據(jù)特定研究要求仔細選擇合適的富集技術至關重要,確保靈敏度、特異性和通量之間的平衡。每種方法都提供了獨特的特點,解決了糖蛋白分析的各個方面,從糖蛋白捕獲的準確性到糖肽回收的有效性。隨著該領域的發(fā)展,這些技術的改進對于提高我們對糖蛋白功能及其在多樣生物場景中重要性的理解至關重要。富集技術在推進糖蛋白質(zhì)組學方面發(fā)揮了關鍵作用,顯著促進了對糖蛋白和IGP的理解和分析。然而,必須承認一個關鍵問題:富集過程不可避免地導致糖蛋白或IGP的部分結(jié)構或組成完整性的損失,這是由于每種方法的偏好。這種損失在一定程度上可能影響糖蛋白質(zhì)組學分析的結(jié)果。為了應對這一挑戰(zhàn),最近的進步集中在開發(fā)聯(lián)合方法上,以更完整或互補的形式分離糖蛋白或IGP,目標是提高IGP分析的準確性和可靠性。
2.4 LC–MS/MS分析
在開始實驗程序之前,選擇適當?shù)姆治龇椒ǎㄙ|(zhì)譜儀、碎裂策略或質(zhì)譜采集方法)對于獲得有意義的結(jié)果至關重要(圖2C)。重要的是,質(zhì)譜儀的分辨率和靈敏度在準確識別IGP中起著關鍵作用。利用不同直徑的色譜柱的色譜技術在這一努力中起著基礎性作用。流速可能受到柱內(nèi)顆粒大小和柱直徑的影響。通常,使用較小直徑的柱子并降低流速可以顯著提高IGP的分離效率。使用與質(zhì)譜兼容的溶劑可以同時啟動超高效或高性能液相色譜(UPLC/HPLC)與質(zhì)譜。此外,反相高效液相色譜(RP-HPLC)技術因其與電噴霧離子化兼容而得到廣泛應用。這種兼容性不僅促進了卓越的分離能力,還確保了一致和可重復結(jié)果的產(chǎn)生。RP-HPLC的獨特優(yōu)勢,包括其出色的分離效率和可靠性,強調(diào)了其在分析化學領域的關鍵作用。
質(zhì)譜發(fā)展之旅既復雜又迷人,標志著科學儀器領域的重要里程碑。在出現(xiàn)的各種技術中,基質(zhì)輔助激光解吸電離(MALDI)質(zhì)譜因其能夠分析糖鏈組成而脫穎而出。這種技術因其高通量和高效率而受到贊譽,成為糖組學領域的基石。另一方面,LC–MS/MS在檢測IGP方面具有更多優(yōu)勢,具有高分辨率和更豐富的糖基化信息。這種復雜的質(zhì)譜分析過程通過三個關鍵步驟展開:電離(將物質(zhì)轉(zhuǎn)化為離子)、質(zhì)量分析(質(zhì)量-電荷比(m/z))和檢測(質(zhì)量分析器和探測器),每個步驟在從樣本到洞察的旅程中都起著至關重要的作用。在最近的進展中,飛行時間(TOF)和(Orbitrap)質(zhì)譜儀已成為臨床糖蛋白質(zhì)組分析的領跑者。這些技術提供了增強的靈敏度和精確度,使它們成為解開臨床樣本中復雜蛋白質(zhì)和糖鏈組成的最佳選擇。
臨床糖蛋白質(zhì)組學領域的最新進展導致了各種串聯(lián)MS/MS碎裂技術和質(zhì)譜采集方法(如數(shù)據(jù)非依賴采集[DIA]、數(shù)據(jù)依賴采集[DDA]等)的出現(xiàn),每種方法都因其產(chǎn)生的不同光譜信息和復雜性而提供了對糖蛋白復雜世界的獨特見解。在這些方法中,逐步碰撞能量/高能量碰撞解離(sceHCD)、電子轉(zhuǎn)移/高能量碰撞解離(EThcD)以及EThcD和sceHCD(EThcD–sceHCD)的組合方法代表了臨床糖蛋白質(zhì)組學中特別有價值的方法。sceHCD,作為N-糖蛋白質(zhì)組學中最常用的技術,能夠從單一光譜分析中產(chǎn)生來自完整N-糖肽的糖鏈和肽鏈的豐富片段離子。然而,當單個序列中存在多個N-糖基化位點時,其在提供識別N-糖基化位點確切位置和N-糖鏈特定組成的清晰光譜證據(jù)方面的能力是有限的。此外,sceHCD不是分析O-糖蛋白質(zhì)組學的首選方法。相比之下,EThcD已成為一種更有效的完整O-糖肽碎裂方法,因為它能夠產(chǎn)生與糖鏈相連的c/z離子,這不僅有助于鑒定肽鏈,還有助于推斷糖基化位點的位置及其糖鏈組成。這種尖端方法顯著提高了識別O-糖基化位點的精度,這一點通過檢測到的片段離子的多樣性和數(shù)量的增加得到了證明。盡管技術進步,但在提高EThcD的效率以更廣泛地應用于糖蛋白質(zhì)組學方面仍存在挑戰(zhàn)。在我們最近的研究中,我們引入了一種創(chuàng)新的混合解離方法,EThcD–sceHCD,它結(jié)合了EThcD和sceHCD的優(yōu)勢。這種整合在糖蛋白質(zhì)組工作流程中形成了一個強大的工具,利用兩種技術的獨特優(yōu)勢。 我們的發(fā)現(xiàn)表明,EThcD–sceHCD顯著增強了對臨床樣本中復雜糖蛋白的分析,如高度糖基化的人類免疫缺陷病毒(HIV)-1 gp120蛋白和Ig。與傳統(tǒng)方法相比,僅依賴EThcD或sceHCD,這種混合方法表現(xiàn)出更優(yōu)越的性能。它提供了更高質(zhì)量的光譜數(shù)據(jù),產(chǎn)生了更詳細的片段離子信息,并識別了更多的完整N/O-糖肽。這些進展強調(diào)了EThcD–sceHCD推動糖蛋白質(zhì)組研究邊界的潛力,為臨床樣本中的糖蛋白提供了更全面和深入的分析。
多樣的碎裂技術在推進我們對健康和疾病中涉及的功能性糖蛋白的理解中發(fā)揮了關鍵作用。這種多樣性使研究人員能夠發(fā)現(xiàn)統(tǒng)一方法無法提供的深入見解。隨著我們深入研究這些MS/MS技術的能力,它們對臨床糖蛋白質(zhì)組學的顯著貢獻預計將豐富我們的理解和為新的研究和臨床應用鋪平道路。盡管如此,數(shù)據(jù)處理,特別是完整N/O-糖肽MS/MS數(shù)據(jù)的注釋,仍然是一個巨大的挑戰(zhàn)。這包括準確識別糖鏈組成和結(jié)構、糖位點和肽鏈骨架。因此,開發(fā)專門的軟件和生物信息學工具非常重要。
2.5 生物信息學分析
精確的分析軟件致力于解碼質(zhì)譜數(shù)據(jù),利用先進的算法。這些算法專門設計用于分析IGP的獨特特征,重點關注提高計算速度和效率。目前,這些軟件工具主要關注分析完整的N/O-糖肽,這與不同疾病的發(fā)生和發(fā)展密切相關。基于核心算法的變化,這些軟件工具主要分為肽優(yōu)先搜索(如Byonic、MSFraggerGlyco等)和糖鏈優(yōu)先搜索(pGlyco系列)。對他們的進展、原理和獨特特征進行了全面的綜述。Byonic被廣泛認為是分析IGP質(zhì)譜數(shù)據(jù)的商業(yè)軟件。該軟件配備了N-糖鏈和O-糖鏈庫以及目標蛋白質(zhì)數(shù)據(jù)庫,可以自動搜索以識別糖蛋白、糖位點和糖鏈組成。該軟件已有效用于分析HeLa細胞中表達的單個糖蛋白的獨特N-糖基化模式,以及尿液糖蛋白質(zhì)組的O-糖基化譜。
MSFragger-Glyco首次引入了開放和質(zhì)量偏移搜索策略。該軟件能夠通過區(qū)分復雜情況下糖肽片段的能量水平來評估混合糖蛋白的能量。因此,它可以分析目標糖肽(能量更高),而忽略糖鏈片段(能量更低),從而減少錯誤。MSFragger-Glyco在解碼多種復雜的N-糖蛋白質(zhì)組和O-糖蛋白質(zhì)組方面表現(xiàn)出色。O-Pair搜索是為分析O-糖基化數(shù)據(jù)而設計的第一個算法。該方法最初使用離子索引的開放搜索與HCD譜圖快速識別肽和O-糖鏈質(zhì)量的配對。61圖形理論方法基于EThcD譜圖中存在的離子定義位點特異性O-糖鏈定位,然后通過磷酸化RS算法的擴展進行定位概率計算。這個算法主要用于定位磷酸化位點,確定假發(fā)現(xiàn)率(FDR),并識別未修飾的肽鏈。GlycoPeptide Finder(GP Finder)專門開發(fā)用于識別廣泛的糖位點。通過分析非特異性蛋白酶消化產(chǎn)生的N-和O-糖肽數(shù)據(jù),可以在保持5% FDR的同時計算消化后肽序列的總體概率。這個工具不僅能夠分析單個蛋白質(zhì)和單個糖位點,還能夠分析未知蛋白質(zhì)混合物。StrucGP專注于識別完整的N-糖肽。它首先通過N-糖鏈的Y離子模式識別肽鏈組分,然后努力通過預定義的子結(jié)構模板解釋糖鏈結(jié)構。此外,它在糖鏈識別后還包括糖鏈級別的質(zhì)量控制措施。
pGlyco系列軟件作為第一個能夠在糖鏈、肽鏈和IGP水平進行質(zhì)量控制的搜索引擎,顯著提高了準確性并加快了IGP匹配。上述提到的軟件工具大大提高了識別完整N-糖肽和O-糖肽的精度。然而,IGP定量分析的軟件工具仍然存在差距。pGlycoQuant通過使用深度學習模型來最小化缺失值,在IGP匹配方面取得了顯著進展。PANDA軟件采用多項式展開技術計算肽元素的自然分布,從而推導出同位素峰的理論相對豐度。隨后,通過在用戶定義的誤差容忍度內(nèi)匹配同位素峰的理論位置,從MS1譜圖中提取觀察到的同位素強度。
上述進展顯著增強了流行的搜索引擎如pGlyco3、Byonic和MSFraggerGlyco的定量能力。然而,這些軟件工具仍然面臨許多需要解決的挑戰(zhàn)。理想的軟件應與各種質(zhì)譜采集方法、不同的碎裂模式、N-和O-糖鏈庫以及不同物種的蛋白質(zhì)庫兼容。開發(fā)能夠有效解碼新糖鏈的糖鏈數(shù)據(jù)庫獨立工具至關重要。能夠同時對多個來源的IGP進行定性和定量分析的軟件的創(chuàng)建,將極大地增強臨床應用和臨床糖蛋白質(zhì)組學的轉(zhuǎn)化。總的來說,當前的軟件工具正在朝著實現(xiàn)更全面的IGP分析、提高準確性和靈活性的方向發(fā)展。
2.6 結(jié)果驗證
基于質(zhì)譜的糖蛋白質(zhì)組學使我們能夠在疾病和對照樣本中發(fā)現(xiàn)數(shù)千種差異表達的糖蛋白、IGP、糖位點和糖鏈。然而,很少有候選糖生物標志物在臨床研究中得到測試。這種現(xiàn)象可以歸因于糖蛋白質(zhì)組學方法的不成熟、缺乏質(zhì)量控制、假陽性檢測、小的臨床樣本量、缺乏定量和驗證實驗,以及在驗證和臨床應用中的挑戰(zhàn)。因此,研究人員應逐漸開始通過必要的結(jié)果驗證實驗來驗證臨床糖蛋白質(zhì)組學發(fā)現(xiàn),類似于臨床蛋白質(zhì)組學,盡管存在挑戰(zhàn)。如圖2E所示,驗證實驗包括分子、細胞和動物實驗。通過這些實驗,可以詳細了解蛋白質(zhì)和糖鏈的結(jié)構和功能。此外,糖蛋白生物標志物的驗證應該進行回顧性和前瞻性,采用多中心獨立樣本驗證方法。驗證結(jié)果的無偏見性是最終評估糖蛋白生物標志物性能的關鍵。
總的來說,臨床糖蛋白質(zhì)組學的方法包括上述幾個程序(臨床樣本選擇、樣本處理、LC–MS/MS分析、生物信息學分析和結(jié)果驗證)。這些方法不斷改進,以提高結(jié)果的質(zhì)量,使我們能夠更深入地理解疾病的糖蛋白質(zhì)組學病理生理學。然而,需要注意的是,這些方法仍然面臨挑戰(zhàn),如無意中選擇不適當?shù)臉颖尽⑹謩訉嶒炦^程、不適當?shù)腗S方法、有限的生物信息學工具和不確定的驗證結(jié)果。重要的是,只有高質(zhì)量的樣本、標準化的樣本處理以及可靠的測量和分析,才對確保實驗的可重復性和可靠性有價值。通過解決這些障礙,臨床糖蛋白質(zhì)組學可以繼續(xù)為我們理解疾病做出重要貢獻。
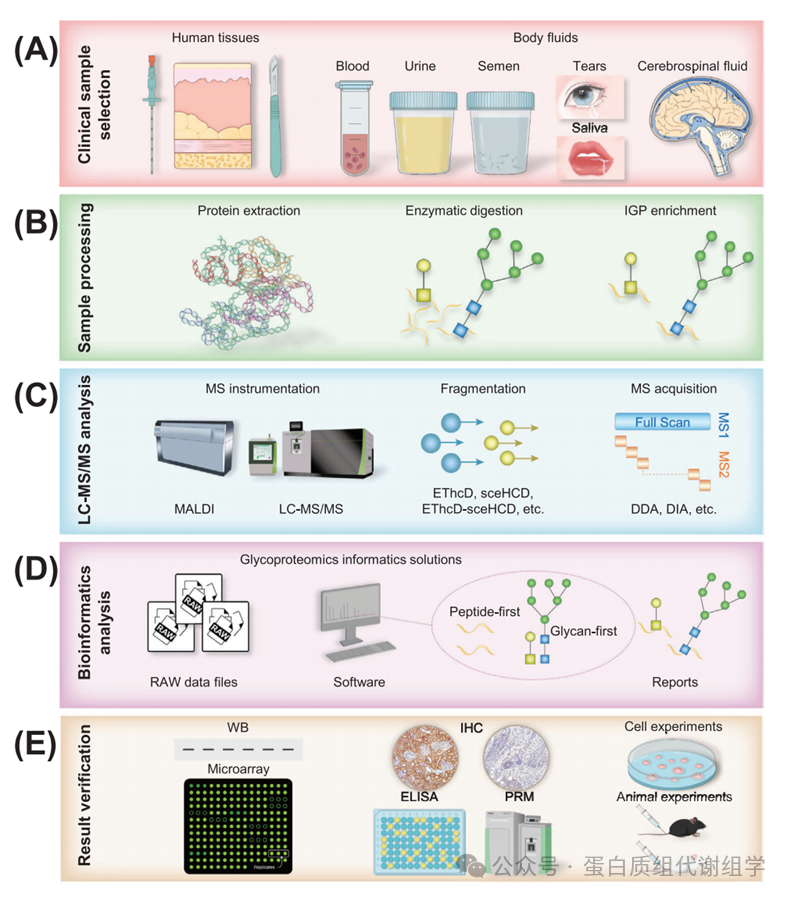
圖2:糖蛋白組學分析的基本流程
- 上一篇:糖肽和糖蛋白分離富集方法精要必看 2024/11/9
- 下一篇:磷酸化蛋白/磷酸化肽段富集與分離方法研究最新進展 2024/11/9